
Multiple sequence alignment
Introduction
The alignment among three or more nucleotides/amino-acids sequences at the same time is known as Multiple Sequence Alignment (MSA), and it is an NP-hard optimization problem. On the one hand, the time complexity of finding an optimal alignment raises exponentially when the input number of biological sequences to align increases. For this reason, the application of parallelism is very interesting. On the other hand, in this work, we deal with a multiobjective version of the MSA problem where the goal is to simultaneously optimize the accuracy and conservation of the alignment.
Multiple Sequence Alignment Problem
Given a set of sequences S: {s1, s2, ..., sk} of lengths |s1|, |s2|, ..., |sk| defined over an alphabet B, e.g. Bprotein={A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y}.
A multiple sequence alignment of S is defined as S': {s'1, s'2, ..., s'k}, where the length of the k sequences is exactly the same. Note that, S' is defined over the same alphabet as S with an additional gap symbol (-).
In this way, a multiple alignment is obtained by adding gaps to the sequences of S so that their lengths become the same. It can be seen as a matrix representation where the rows are sequences and the columns represent aligned symbols. Each column of an alignment must contain at least one symbol of B, in other words, a column with all gaps is not allowed. An example of multiple sequence alignment may be:


In this work, we propose the use of multiobjective metaheuristics for solving this NP-complete optimization problem, searching the best solution that simultaneously maximizes the weighted sum-of-pairs function with affine gap penalties (WSP, f1) and the number of Totally Conserved (TC, f2) columns score. Therefore, we have two objective functions for maximization.
On the one hand, the weighted sum of pairs with affine gaps (WSP, f1) can be calculated by using the following equations:
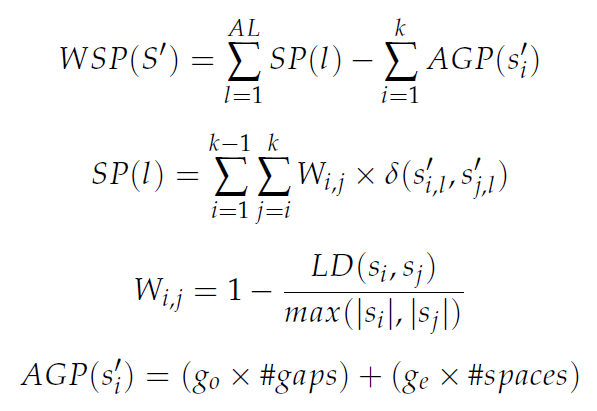
In the previous equations, AL is the alignment length, SP(l) is the sum-of-pairs score of the lth column, delta is the substitution matrix used, either Pointed Accepted Mutation (PAM) or Block Substitution Matrix (BLOSUM), Wi,j refers to the sequence weight between sequence si and sj, LD refers to the Levenshtein Distance between two non-aligned sequences (that is, the minimum number of insertions, deletions or substitutions required to change one sequence into the other), AGP(s'i) is the affine gap penalty score of sequence s'i, go is the weight to open the gap and ge is the weight to extend the gap with one more space.
On the other hand, the number of Totally Conserved (TC, f2) columns score is defined as the number of columns that are completely aligned with exactly the same compound. This second objective function needs to be maximized to ensure more conserved regions within the alignment. In the previous example, the symbol '*' indicates a column completely aligned with exactly the same compound, therefore, in that example f2=4.
Note that we have constrained the maximum number of columns (alignment length) to be at most 50% longer than the longest sequence in the unaligned set of sequences (input sequence set). This choice was based on the observation that solutions to common alignment problems rarely contained more than 50% gaps.
References
- "A Characteristic-based Framework for Multiple Sequence Aligners". Álvaro Rubio-Largo, Leonardo Vanneschi, Mauro Castelli, Miguel A. Vega-Rodríguez. IEEE Transactions on Cybernetics, Volume 48, Issue 1, IEEE, Piscataway, NJ, USA, 2018, pp. 41-51, ISSN: 2168-2267. DOI: 10.1109/TCYB.2016.2621129. (Impact factor = 10.387, Quartile Q1)
- "A Parallel Multiobjective Metaheuristic for Multiple Sequence Alignment". Álvaro Rubio-Largo, Mauro Castelli, Leonardo Vanneschi, Miguel A. Vega-Rodríguez. Journal of Computational Biology, Volume 25, Issue 9, Mary Ann Liebert, New Rochelle, NY, USA, 2018, pp. 1009-1022, ISSN: 1066-5277. DOI: 10.1089/cmb.2018.0031. (Impact factor = 0.879, Quartile Q3)
- "Multiobjective Characteristic-based Framework for Very-Large Multiple Sequence Alignment". Álvaro Rubio-Largo, Leonardo Vanneschi, Mauro Castelli, Miguel A. Vega-Rodríguez. Applied Soft Computing, Volume 69, Elsevier Science, Amsterdam, The Netherlands, 2018, pp. 719-736, ISSN: 1568-4946. DOI: 10.1016/j.asoc.2017.06.022. (Impact factor = 4.873, Quartile Q1)
- "Swarm Intelligence for Optimizing the Parameters of Multiple Sequence Aligners". Álvaro Rubio-Largo, Leonardo Vanneschi, Mauro Castelli, Miguel A. Vega-Rodríguez. Swarm and Evolutionary Computation, Volume 42, Elsevier Science, Amsterdam, The Netherlands, 2018, pp. 16-28, ISSN: 2210-6502. DOI: 10.1016/j.swevo.2018.04.003. (Impact factor = 6.330, Quartile Q1)
- "Reducing Alignment Time Complexity of Ultra-large Sets of Sequences". Álvaro Rubio-Largo, Leonardo Vanneschi, Mauro Castelli, Miguel A. Vega-Rodríguez. Journal of Computational Biology, Volume 24, Issue 11, Mary Ann Liebert, New Rochelle, NY, USA, 2017, pp. 1144-1152, ISSN: 1066-5277. DOI: 10.1089/cmb.2017.0097. (Impact factor = 1.191, Quartile Q2)
- "Using Biological Knowledge for Multiple Sequence Aligner Decision Making". Álvaro Rubio-Largo, Leonardo Vanneschi, Mauro Castelli, Miguel A. Vega-Rodríguez. Information Sciences, Volume 420, Elsevier Science, New York, NY, USA, 2017, pp. 278-298, ISSN: 0020-0255. DOI: 10.1016/j.ins.2017.08.069. (Impact factor = 4.305, Quartile Q1)